Our latest focus is trying to bring data to bear on common questions we get asked by clients. Last month the topic was: how well does my device need to perform to get premarket clearance from FDA? This month it is: how big does my sample size need to be for any necessary clinical trial for premarket clearance?
To date, our typical answer has been, it depends.[1] We then explain that it’s not really a regulatory question, but a question for a statistician that is driven by the design of the clinical trial. And the design of the clinical trial is driven by the question the clinical trial is trying to answer. Persistent clients with an MBA might say, well give me an estimated range because it’s a huge driver of the cost of obtaining FDA clearance and we need a reasonable estimate upfront before we design the trial. Hopefully, the results of this month’s post will help with that discussion, although the precise answer will still depend tremendously on the nature and purpose of the clinical trial.
Caveat
Last month, Brad started with a huge caveat before he even presented the data. For that post[2] Brad obtained the data by using natural language processing on posted 510(k) summaries. He pointed out the infirmities of that approach.
The good news is this month we have a more reliable, structured data set taken from clinicaltrials.gov. The bad news is, the data are still incomplete, and we fear significantly so. The last time Brad used a data set from clinicaltrials.gov, Brad explained in a February 1, 2022 post[3] the regulatory history of that website. Brad’s own observation is that the medical device companies were relatively slow in recognizing the need to register their clinical trials on that website. As a result, we fear that especially in the early years the data were incomplete.
In trying to think through how that incompleteness might bias the data, while we have no data on this, we fear that companies doing smaller scale clinical trials less likely to conclude that their trials qualified as “official” clinical trials requiring registration on the website. It seemed more typical that the larger the trial, the more likely the company was to register it. We can’t prove that. It’s merely a theory based on personal experience.
But again, as you look at the results of this analysis, please have your eyes wide open.
Findings
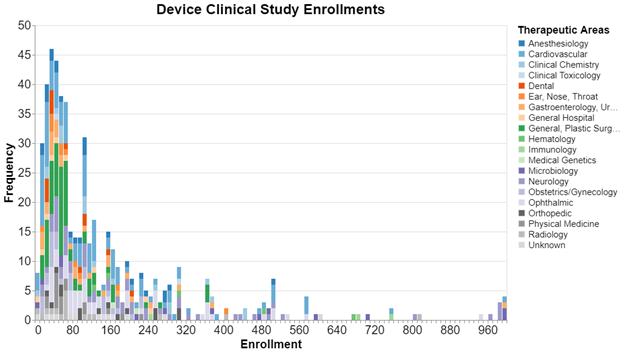
Here, sorted by the sample size in bins of 10 are the clinical trials submitted by device sponsors to clinicaltrials.gov since 2013. We cut off the sample size at 1000 because it got even more sparse above that. The color coding shows the therapeutic area in which the trial was conducted.
Data Set Used
For those of you who may be unfamiliar with clinicaltrials.gov, the Final Rule for Clinical Trials Registration and Results Information Submission (42 CFR Part 11) clarified and expanded the regulatory requirements and procedures for submitting registration and results information for certain clinical trials to ClinicalTrials.gov, in accordance with Section 801 of the Food and Drug Administration Amendments Act (FDAAA 801). The Final Rule has been in effect since January 18, 2017.
In defense of those companies that have been confused about this, much of what is written on official clinicaltrials.gov website is anything but clear. For example, here’s a passage:
Registration is required for studies that meet the definition of an "applicable clinical trial" (ACT) and either were initiated after September 27, 2007, or initiated on or before that date and were still ongoing as of December 26, 2007. ACTs, as defined in section 402(j) of the PHS Act, include the following: …
- Certain studies of FDA-regulated medical devices, excluding small clinical trials to determine feasibility and certain clinical trials to test prototype devices, but including FDA-required pediatric postmarket surveillancesof a device product.[4]
If you’re like us, you immediately want to know what “certain” studies means, but the text that follows does not explain. Instead, the text simply refers you back to the regulation 42 CFR 11.10 and goes on to say:
ACTs generally include interventional studies (with one or more arms) of FDA-regulated drug, biological, or device products that meet one of the following conditions:
- The trial has one or more sites in the United States
- The trial is conducted under an FDA investigational new drug application or investigational device exemption
- The trial involves a drug, biological, or device product that is manufactured in the United States or its territories and is exported for research
Again, the language says “generally”, but doesn’t explain the equivocation. Instead, the website refers the reader to yet another resource as follows:
For complete statutory definitions and more information on the meaning of Applicable Clinical Trial, see the Checklist and Elaboration for Evaluating Whether a Clinical Trial or Study is an Applicable Clinical Trial (ACT) ("ACT Checklist"), which follow the criteria specified in 42 CFR 11.22(b), to determine whether a study initiated on or after January 18, 2017, is an ACT subject to the expanded registration requirements under the Final Rule.
That checklist is a nine-page document full of detailed definitions. The easiest way to summarize it is to say that all of those interventional clinical trials that must be conducted under an Investigational Device Exemption or IDE and are not a device feasibility study must register on the clinicaltrials.gov site. As you may know, a device must be investigated under an IDE granted by the FDA if it involves significant risk. A device feasibility study is less clearly defined, but it does seem clear that a feasibility study is not one that will be submitted to FDA in support of a marketing application.
That scope is still confusing, because “conducted under an IDE” is not the same thing as “requiring FDA approval of an IDE.” As the checklist itself points out “Device products that are considered to be subject to section 510(k), 515, or 520(m) of the FD&C Act include significant risk devices for which approval of an IDE is required under section 520(g) of the FD&C Act or non-significant risk devices that are considered to have an approved IDE in accordance with 21 CFR 812.2(b). [Source: 81 FR 65012]”
What exactly is the website trying to say in that passage? Is the website suggesting that any device subject to an IDE, whether it requires FDA approval or not, must register?
When completely confused, we go back to the regulation itself 42 CFR 11.22(b)(1) which states in part that the following studies must be registered:
(ii) The study is a clinical trial with one or more arms that meets all of the following criteria:
(A) Study Type is interventional;
(B) Primary Purpose of the clinical trial is other than a feasibility study;
(C) The clinical trial Studies a U.S. FDA-regulated Device Product; and
(D) One or more of the following applies:
(1) At least one Facility Location is within the United States or one of its territories,
(2) A device product under investigation is a Product Manufactured in and Exported from the U.S. or one of its territories for study in another country, or
(3) The clinical trial has a U.S. Food and Drug Administration IDE Number.
Actually, we find the regulation clearer than anything the government has written about it. To need to register, there must be an IDE number. IDE numbers are assigned by FDA as a product of an application submitted to the agency. Thus, IDE's that do not require an FDA IDE submission do not get assigned an IDE number.
Thus, as a general proposition, this data set should include at least information on studies submitted to FDA for clearance or approval for devices the use of which involves significant risk. But you can also see why there might be some confusion about which device studies are required to register. Further, it is important to note that other studies can be registered even if they’re not legally required to be.
FDA requests that companies submit in their marketing applications information showing their compliance with that registry in their application for premarket clearance.[5]
The bottom line is that we think this data set is incomplete but in an idealized way should include larger clinical trials (i.e., not feasibility trials) done for the purpose of securing FDA clearance for medical devices the study of which produces significant risk.
Methodology
In comparison to other posts Brad has done, the methodology here is reasonably simple. We started on the FDA website and used the 510(k) database to identify all the NCT numbers (these are the numbers assigned by clinicaltrials.gov) for clinical trials submitted in connection with a 510(k). We found 626 such clinical trials since 2013. By the way, this was harder than it should have been because we needed to use the FDA's 510(k) database, instead of openFDA where the agency is supposed to share its data. The agency hasn't gotten around to sharing this on the openFDA site.
We then used that list of NCT numbers to query the clinicaltrials.gov site to extract the information provided by sponsors. This information included the sample size. We then went back to the FDA site to extract from the 510(k) data the clinical panel assigned to each related submission.
Interpretation
The big take away is the skew toward the smaller end of the sample size. For these trials, it's quite common for a study to have fewer than 100 subjects. Regarding therapeutic area, this is especially true for the greens and oranges. There are very few green or orange bars over 100 subjects. We did not run similar numbers for drug studies, but drug studies tend to be much higher in terms of the number of enrolled subjects.
We should stress that these data are about the sample size of a particular study. It’s not uncommon, in fact it happens about 25% of the time, that sponsors submit two or more studies in support of a given submission. As a result, we cannot say that this is the total sample size for all studies required for FDA clearance.
It’s important to point out that all these studies are from premarket notification submissions, and not from premarket approvals. We would get different results if we focused on class III medical devices that required premarket approval.
We are quite honestly a bit unsure how to extrapolate from these data the likely sample sizes for clinical trials submitted for 510(k) clearance in connection with nonsignificant risk devices. Sometimes the fact that a device study involves significant risk means that the numbers need to be kept to a minimum to avoid unduly subjecting subjects to risk. But at the same time, with devices that involve significant risk, the sample size still needs to be large enough to give the FDA confidence that the product is indeed safe and effective. On the whole, we suspect data for nonsignificant risk devices may look somewhat similar to these, because fundamentally the design of the trials will be similar.
Another area for speculation is clinical trials exempt from IDE requirements perhaps because they involve in vitro diagnostic devices or IVDs. Such studies typically would not be registered on clinicaltrials.gov and thus not in the summary above. FDA guidance for IVD devices appropriately suggests that sample size be driven by a statistical considerations.[6] Unfortunately, we do not have organized data on such trials to compare with the results above, but it’s likely that they would be significantly different because of the different risk considerations, purposes and design of the trials.
Conclusion
Clients typically find it reassuring that it is common that a clinical trial for a device might include fewer than 100 subjects. Keep in mind that these are significant risk devices, and therefore do not include nonsignificant risk devices or IVD’s. The data also don’t include clinical trials submitted for PMA approval, and in about 25% of the cases a submission requires two or more clinical trials. All that said, these data are typically encouraging for small device startups without excess capital because they show that the clinical trials necessary for FDA clearance do not typically need to enroll large numbers of participants.
* * * *
The Unpacking Averages® blog series digs into FDA’s data on the regulation of medical products, going deeper than the published averages. The opinions expressed in this publication are those of the author(s). Subscribe to this blog for email notifications.
ENDNOTES
[1] https://www.greenlight.guru/blog/8-tips-before-calculating-sample-size-medical-device-studies
[2] https://www.healthlawadvisor.com/2023/09/05/unpacking-averages-how-accurate-do-class-ii-medical-devices-need-to-be-to-obtain-510k-clearance/
[3] https://www.healthlawadvisor.com/2022/02/01/unpacking-averages-assessing-the-racial-composition-of-drug-clinical-trial-subjects/
[4] https://classic.clinicaltrials.gov/ct2/manage-recs/fdaaa
[5] https://www.fda.gov/science-research/clinical-trials-and-human-subject-protection/fdas-role-clinicaltrialsgov-information
Blog Editors
Authors
 Board of Directors / Member of the Firm
Board of Directors / Member of the Firm