Recalls have always been a bit of a double-edged sword. Obviously, companies hate recalls because a recall means their products are defective in some manner, potentially putting users at risk and damaging the brand. They are also expensive to execute. But a lack of recalls can also be a problem, if the underlying quality issues still exist but the companies are simply not conducting recalls. Recalls are necessary and appropriate in the face of quality problems.
Thus, in terms of metrics, medical device companies should not adopt as a goal reducing recalls, as that will lead to behavior that could put users at risk by leaving bad products on the market. Instead, the goal should be to reduce the underlying quality problems that might trigger the need for recall.
What are those underlying quality problems? To help medical device manufacturers focus on the types of quality problems that might force them to conduct a recall, we have used the FDA recall database to identify the most common root causes sorted by the clinical area for the medical device.
Background
For most recalls, FDA reviews the documentation and perhaps conducts an on-site inspection to reach a conclusion about what the root cause is for the needed recall. They do this at least in part because they want to make sure that the manufacturer is appropriately focused on the correct root cause, both so that the manufacturer conducts the recall properly as to scope and communications, but also so that they fix the real underlying problem.
According to Chapter 7 of FDA’s Investigations Operations Manual on recall activities, FDA examines root causes for the following reasons:
7.2.2 – ROOT CAUSE INSPECTIONS
If FDA learns of a potentially violative product that may cause or has caused a class I or significant class II recall, an inspection may be assigned to determine the root cause(s) of the problem(s). Deficiencies in the firm’s corrective and preventive action should be documented as violations subject to possible regulatory action. An important objective of the inspection is to identify the root cause for the recall and assure the firm has implemented effective corrective actions to eliminate its recurrence. In some cases, firm management will have conducted its own analysis and reached conclusions about the problem and its root cause. It is important to verify that the firm’s conclusions and judgments, about the root cause of the problem that led to the recall, are discriminating enough to identify the true cause(s) and steps taken are sufficient in depth and scope. Without identifying the true root cause, it will be difficult for the firm to implement an effective corrective action.
In looking at the data set, it appears that FDA tries to arrive at a conclusion regarding root cause for just about every recall.
It’s important to note that FDA’s recall database is reasonably complete with regard to class I and class II recalls, which are the most serious. Manufacturers are not required to report class III recalls (the least serious recalls), and many don’t.
Visualization
I chose to use a heat map because I wanted to convey the frequencies of the various root causes by the range of medical specialties in which different devices are classified. In presenting the data, I eliminated one medical specialty, “medical genetics,” because there were so few reported. I also deleted the catchall categories for root causes, for example “other” and “under investigation,” because they didn’t reveal anything useful.
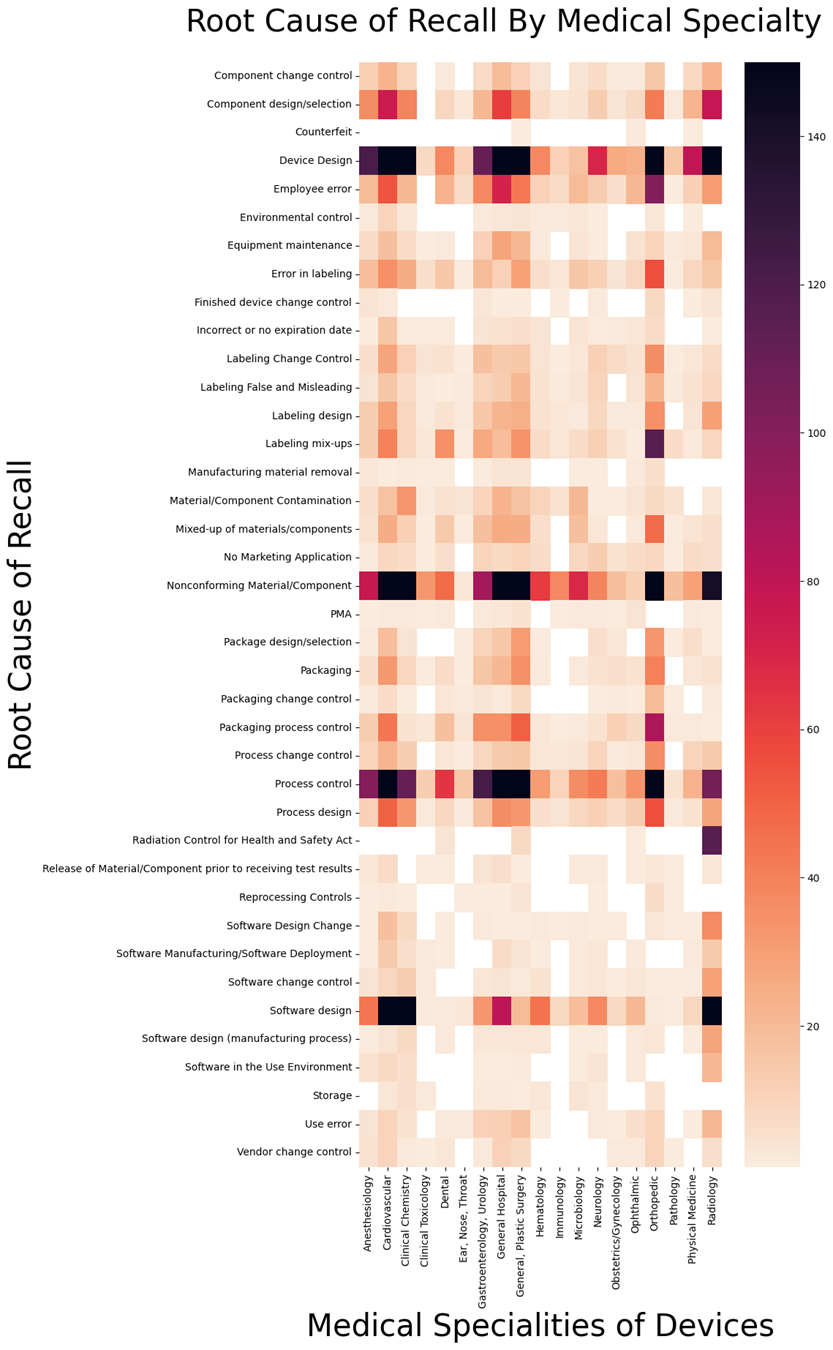
This heat map shows everything from one recall to 150 in each category. I capped it at 150 to more clearly differentiate the lower end of the spectrum. There were numerous outliers that were well over 150, but it didn’t seem worth skewing the data to show those specifically.
Methodology
FDA maintains a database of all recalls since 2003, so I filtered just those involving medical devices. That produced roughly 46,000 recalls. However, although they are technically different recalls, many of these recalls are simply redundant with previously reported recalls as firms expand the number of devices that are subject to the recall. Technically, each time a company expands a recall, the new information gets entered as a separate recall. But from a root cause standpoint, it seems to me that’s meaningless duplication or noise.
For each recall, FDA assigns a “res_event_number,” a five digit, numerical designation to a specific recall event used for tracking purposes. This number stays the same even as manufacturers add additional products to an existing recall. I felt it was more accurate to simply portray the number of root causes by this event number to avoid double counting. After removing duplicates, there were a little over 18,000 unique recalls from 2003-2022 (spring).
To create the visualization, I used FDA’s “root_cause_description,” the general type of recall cause. Per FDA policy, recall cause determinations are subject to modification up to the point of termination of the recall. All but seven medical device recalls in the database had a root cause assigned to them. Most of them are self-explanatory. The root cause that is labeled “PMA” means that the product had no FDA approval. They just say in shorthand “PMA,” even though technically the product might qualify for a 510(k) premarket notification.
Interpretation
Certain factors jump out from the heat map. For example, the root causes of “device design,” “nonconforming materials,” “process controls,” and “software design” tend to explain quite a few recalls across all clinical specialties.
On the other hand, as to be expected, some of the more unique requirements such as “radiation control” only apply to certain devices.
If you look outside those obvious things, there are interesting nuances such as, why are labeling issues so prevalent in orthopedic recalls? Indeed, look generally at the labeling type problems and see how prevalent they are in orthopedics versus other areas. I also wonder why employee errors are so prevalent in orthopedics, but virtually nonexistent in clinical toxicology.
In the area of software design, I find it interesting that cardiology has such trouble in that area, where pathology does not. While pathology has historically been filled with visual instruments, I would have assumed the movement to digital pathology would lead to more software issues. But so far it really hasn’t.
It’s also curious that radiology struggles with component design, but clinical toxicology doesn’t.
Conclusion
Knowing the story behind these numbers is essential to properly interpreting them. Thus, my hope is that you will read these in light of your knowledge about your particular part of the industry and that the data will provide some insight regarding where your company should focus its quality efforts. I think the relative differences between clinical specialties reveals potential areas of focus that might be productive, if you have the background context to interpret them.
* * * *
The Unpacking Averages® blog series digs into FDA’s data on the regulation of medical products, going deeper than the published averages. The opinions expressed in this publication are those of the author(s). Subscribe to this blog for email notifications.