You might be thinking, that’s an odd title: obviously FDA’s breakthrough device designation is helpful. However, after looking at the data, my conclusion is that I would avoid the breakthrough device designation for any product that qualifies for the 510(k) process. The process is likely not helpful for such devices.
[Update - August 3, 2022: See the bottom of this post.]
Background
Why would I even question whether the program is helpful? Well, to be honest, in my 35 years of working with medical device companies, the program runs counter to everything I typically try to do in a device submission. My goal in nearly every device submission is to try to get through the FDA process with as little attention from FDA as possible.
FDA summarizes the benefits of the program this way:
The Breakthrough Devices Program offers manufacturers an opportunity to interact with the FDA's experts through several different program options to efficiently address topics as they arise during the premarket review phase, which can help manufacturers receive feedback from the FDA and identify areas of agreement in a timely way. Manufacturers can also expect prioritized review of their submission.
When young people read that, they might think, wow, great! Free advice! But when a seasoned person looks at it who has experience with FDA, their first thought is, do I really want FDA more involved in the process?
FDA is not a taxpayer subsidized consulting organization. They are a regulatory body whose mission to protect patients. They are a law enforcement agency. They are not trained in constructive problem-solving of the type that a consultant would do. That’s not a criticism. In my opinion, FDA is very good at what it does: regulating. People can’t be good at everything, but also there is a bit of a conflict between the mission of protecting patients and the objective of finding or discovering the least burdensome pathway through the agency.
If you get lots of attention from FDA, you could quite easily get lots of very helpful suggestions such as:
- We would like you to do a clinical trial.
- We would like the clinical trial you are planning to be three times larger.
- We would like you to improve the rigor of the design of the clinical trial you are planning.
- We would like you to do a much more comprehensive risk assessment that includes a variety of risks that cause us concern.
My point is that FDA doesn’t just flip into becoming a regulatory consultant for breakthrough devices offering only helpful suggestions that the manufacturer will like. The breakthrough program just makes the agency more involved in the upfront decision-making about what kind of evidence they want to receive.
Unfortunately, we can’t evaluate perhaps the biggest part of the breakthrough program which is the effect that the program has on the development time of the medical device leading to a regulatory submission. There is just no data on that. And that is the biggest claim that FDA makes is that they will cause the development process to be more efficient.
I suppose in a sense the process is more efficient through FDA’s eyes because they communicate more quickly and more clearly the increases in the evidence that they expect to receive. But I’m also here to tell you that if the company takes on more of the up-front decision-making about what evidence it plans to develop and submit, and keep the agency out of such planning, the company can come up with more modest evidence that FDA will accept.
The part of FDA’s claim that we can test is the following: “Manufacturers can also expect prioritized review of their submission.” While until recently we had no ability to evaluate that claim because there was no public data set for the review of breakthrough devices, this spring FDA for the first time released a list of submissions that were subject to the breakthrough device designation.
To be sure, though, the list is not comprehensive with regard to all devices that have entered the breakthrough device program. FDA explains that: “As of March 31, 2022, CDRH and CBER have granted 657 Breakthrough Device designations.” Those designations have been granted over a number of years, as illustrated by the following FDA graph:
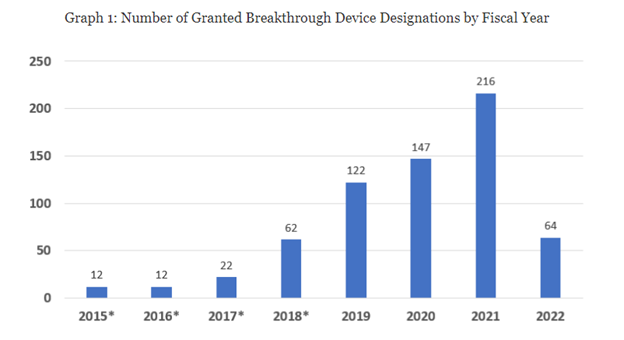
While FDA has made over 600 breakthrough device designations, the database that they released has only 44 marketing authorizations that involve the breakthrough device designation. That statistic by itself is very concerning. Think about it: they’ve granted over 600 breakthrough device designations but they have only 44 that the agency has cleared or approved. While many of the designations are recent, meaning from 2019 forward, we still would expect more by now. This statistic raises a fundamental concern that these breakthrough devices are not even getting cleared or approved.
But even though it’s only a small data set, the data give us some ability to evaluate the impact of the breakthrough designation on the actual review times for those products.
Findings
While I was already suspicious, the findings surprised even me.
- For those breakthrough devices submitted through the 510(k) process, the breakthrough devices took three days longer to be cleared than comparable devices not in the breakthrough device designation category.
- For those breakthrough devices submitted through the de novo process, the breakthrough devices received a final decision 75 days quicker than the average for all products submitted through the de novo process.
To be clear, those numbers are based on few data points as already observed. But those are all the data we have.
Methodology
Obviously, the objective was to be as fair as possible. I recognize that breakthrough devices by their very nature are more likely to be cutting-edge products technologically, and more likely to deal with serious diseases or conditions, so I wanted to come up with a methodology that took that into account as best as possible.
For devices that go through the 510(k) process, due to the elevated novelty and risk, I did not want to compare the speed by which they were reviewed with the speed generally for all 510(k) devices. That would ignore the heightened risk profile.
As you undoubtedly know, devices that are subject to the 510(k) process are put into product codes that are specific to the technology and indications involved. If you’re a regular reader of this blog post, you will know from analysis last year that there’s wide variability across product codes regarding review times. As a result, even though the method involves shrinking the data set substantially, I focused in comparing the breakthrough device review times on the corresponding product code specific review times in which those breakthrough devices were reviewed.
There were only 11 devices in the breakthrough program that have been cleared, so admittedly this is a small data set. While it is small, from a statistical standpoint it’s not a sample. It is purportedly the universe of such devices as of March 1, 2022, although it wouldn’t surprise me if FDA released an incomplete data set. Further, in looking at the raw data for these breakthrough devices compared to product code specific review times, the individual differences in speed to decision is highly variable with some breakthrough devices being cleared much after the average for a given product code, while others are cleared much faster.
For devices that are subject to the de novo process, as you know, that process creates a product code rather than placing a device into an existing product code. As a result, there is no real way to compare first of a kind device with other device review times because, well, they are first of a kind. Thus, for the breakthrough devices reviewed in the de novo process, I simply picked the overall average de novo review time as the comparator.
I would note, though, that the de novo review times are highly variable and so there must be factors not captured here that influence how quickly a particular product is reviewed through the de novo process. Again, while the time savings are 75 days on average, the actual de novo decision time savings are highly variable within the breakthrough device category.
Conclusion
On the whole, acknowledging the limitations that I stated such as the high degree of variability, small data size and the lack of any way to review the claim that the development process is faster, I would have to seriously question the value of the breakthrough device designation for devices that qualify for a 510(k). If I were going to take the risk that FDA would tell me to develop much more expensive data than I had planned for a breakthrough device, I would want at least a more compelling case that the review time should be shorter for those that qualify.
On the other hand, for devices that need to go through the de novo process, I do think the data support the value of obtaining the breakthrough device designation. While a quick review certainly is not a foregone conclusion given the variability of the data, on average it seems like a good bet.
There is an intuitive support for this conclusion as well, in that if the device can fit into the 510(k) process, I would have to seriously question even the conceptual value of the breakthrough device designation program. But for devices that are truly unique and thus requiring the de novo process, the streamlined review process is clearly more appealing.
Update - August 3, 2022
I don't know if it was spurred by my blog post, but just today FDA released data for the third quarter of the fiscal year with regard to the breakthrough device designation program. I've done the analysis of the newly released data.
FDA reports that as of June 30, 2022:
- There have now been a total of 693 devices granted breakthrough device designation.
- There are now a total of 54 marketing authorizations, up from the previously reported 44 marketing authorizations at the end of the prior quarter. In other words, 10 new marketing authorizations for the quarter.
Recalculating the averages,
- For the 510(k), instead of three days slower for breakthrough devices, it's now about one half day slower for breakthrough devices. Essentially dead even.
- For de novo, instead of a 75 day advantage for breakthrough devices, it's down to a 64 day advantage for breakthrough devices.
The new data and the new averages don't change any of the conclusions in this post.
* * * *
The Unpacking Averages® blog series digs into FDA’s data on the regulation of medical products, going deeper than the published averages. The opinions expressed in this publication are those of the author(s). Subscribe to this blog for email notifications.