This month’s post focuses on how timely FDA decisions are in categorizing new diagnostics under the Clinical Laboratory Improvements Amendments of 1988 (CLIA). The answer is that, on average, the agency does okay, but they also sometimes may miss their own guideline by a wide margin. I use the word “may” there because the FDA data set is inadequate to support a firm conclusion. I’ll explain more about that below, but this is another case of FDA releasing incomplete data that frustrates data analytics.
As those in the laboratory diagnostics field know, FDA has responsibility for categorizing new diagnostics as either high complexity, moderate complexity, or waived due to the simplicity of the test. These categorizations determine which laboratories can perform the tests, as laboratories have to be certified as capable of performing a certain level of test complexity. Thus, the categorization process plays a key role in determining which laboratory customers can use a given test.
If an in vitro diagnostics company wants a laboratory test placed in the waived category, they can submit an application to FDA requesting that categorization, either independently or together with a premarket submission such as a 510(k). Those waiver applications are subject to user fees, and consequently there are metrics for the review of those applications that FDA is expected to meet. FDA tracks those metrics and publishes periodic updates on their performance. As a consequence, in this post I’m not going to address those.
Instead, because there are no published metrics by FDA on how the agency does in its review of new laboratory tests that are not subject to a waiver application—diagnostic tests that are supposed to be reviewed and categorized automatically after FDA clearance or approval—I focus on those. In FDA’s vocabulary, the decision process for these is referred to as a CLIA Record, or “CR” for short.
The process for FDA to review and categorize these tests is defined in a 2017 FDA guidance entitled, “Administrative Procedures for CLIA Categorization.” There are actually two different processes through which FDA might categorize a test. The first is described in II.A. of that guidance, and FDA refers to these as “concurrent.” They got that name because FDA conducts the review at the same time they review the premarket submission. On page 6 of that guidance, FDA says that it “will attempt to notify sponsors of the complexity categorization within two weeks of a positive marketing decision (e.g. a substantial equivalence determination for a 510(k) or approval decision for a PMA).”
The second type of categorization decision is referred to as “standalone,” and is described in II.B. of the guidance. It is standalone in the sense that the categorization decision follows a request by the IVD company, rather than being automatically connected to a premarket decision. A manufacturer might make this request, for example, for an exempt device or where a device has been changed, but not so much that it requires a new 510(k).
I chose to focus this analysis on the concurrent reviews to see whether FDA does indeed meet the two week standard for making those decisions. The answer is: most of the time they do, but apparently not always. In some cases, they appear to miss their deadline by several multiples. But again, I need to point out that the FDA data set is defective in some ways, and I explain more about that in the limitations section below.
Chart
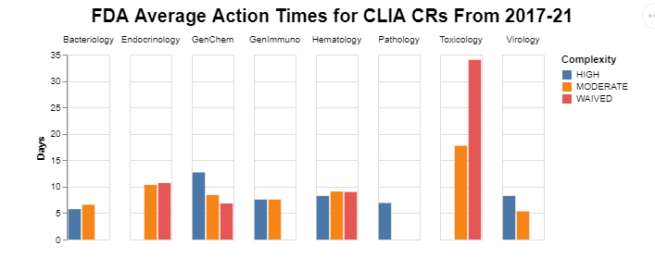
To unpack the averages, I first wanted to see whether there was variability in FDA action times across the different types of analytes, broken down by test complexity.
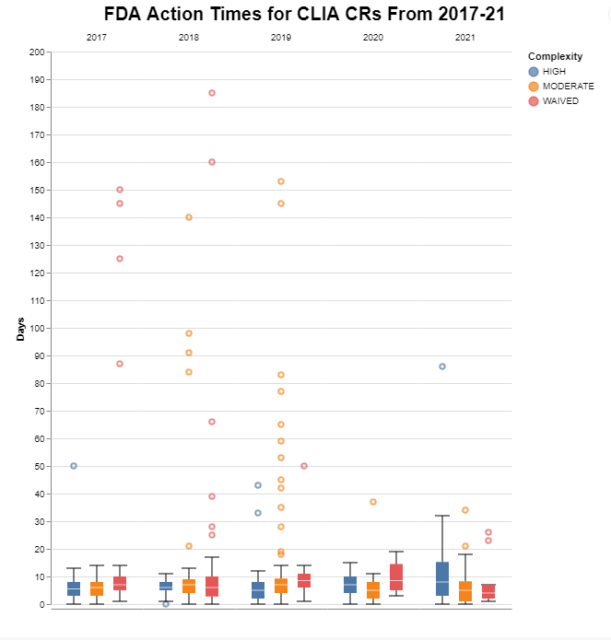
I also wanted to visualize the raw data (as opposed to the averages presented in the first chart) to see whether there was variability across the years, again broken down by test complexity, and whether there were outliers. In these box plots, if you’re not familiar with that type of visualization, the actual boxes represent the middle two quartiles of the data, with the white line representing the median. I set the stems on the top and the bottom to represent the conventional distance of 1.5 times the height of the box. This is just a convention that shows typically where we would expect the top quartile and the bottom quartile to be, but then I added the actual raw data that are outside of that conventional distance to depict the outliers.
Methodology
FDA releases through its website data on its CLIA categorization decisions. In that data set, in addition to identifying the categorization decision, the agency identifies the application number of what’s called the Parent marketing submission. As a consequence, I took the CLIA data for the five calendar years 2017 through 2021 and joined it to the clearance data released through FDA’s openFDA portal. By doing that, I was able to get the dates for both the categorization decision as well as the clearance decision. So I’m just comparing those two dates in the chart above.
But I also do some filtering in order to focus on those areas in which I am most interested. First, I am only interested in those categorizations decisions that are made automatically (i.e. the concurrent decisions), not those made as a result of an application for waiver. There typically are only a handful of applications for waiver anyway in a given year, but I excluded those because they are subject to FDA’s own published analysis.
Next, I want the data to be relatively homogenous in terms of risk, so I chose to focus on the device submissions which are 510(k)’s, as opposed to premarket approval applications and biological device applications, as well as de novo decisions. So I excluded everything but 510(k)’s. That excludes instances where the laboratory test was exempt from FDA review altogether. FDA procedures give the agency 30 days to review those requests for categorization.
For these purposes, with regard to 510(k)’s, as already explained, FDA makes two different kinds of decisions – concurrent and standalone. Unfortunately, in the data FDA presents, the agency doesn’t distinguish between those two different actions. For my purposes, though, they represent completely different issues and I wanted to focus on only the concurrent decisions. That required some creativity on my part.
This isn’t perfect, but I reasoned that the first decision on a particular 510(k) would likely be the original decision (or in FDA’s vocabulary, the concurrent decision), and any subsequent decisions referencing that same 510(k) would likely be a labeling or other update (a so-called standalone decision). So I filtered the data set to only look at the first of any multiple CLIA actions on a single 510(k). Even though I only wanted to look at the last five years, that meant I looked at a 10 year data set in order to capture earlier clearances to filter out subsequent labeling decisions.
That didn’t work perfectly, because when I looked at the data after filtering it, there were still quite a few decisions that look like it’d taken a year or even up to five years for the agency to make. Here I gave the agency the benefit of the doubt, and assumed that those were standalone decisions, but that for some reason the data set didn’t include the earlier concurrent decision. As a result, I simply filtered out every remaining decision that was over 300 days as well as any submission where the 510(k) was more than 10 years old. Three hundred was somewhat arbitrary, but when I looked at the data, there seem to be a natural break there.
And finally, these submissions cover a range of analyte categories. Since I’m dealing with averages, I wanted to make sure that I only used categories of analytes that had an adequate number of submissions to give a meaningful average. So I filtered out all of those analyte categories that didn’t have at least five applications over the five years. That sounds relatively arbitrary, but when I looked at the data, there was a natural break there between the very small and the much more numerous.
I included the bars for high complexity, in that they show how long FDA takes to make a decision, even if the decision is simply to put it in high complexity. For background, though, the default category is high complexity. Thus, laboratories qualified to run high complexity tests can run those tests and frankly any others while waiting for an FDA categorization decision.
Limitations
As I explained in the methodology, I was frustrated by the number of significant outliers in the data. I also wanted to see if there was some better, more reliable way to distinguish between concurrent and standalone decisions. So I contacted FDA.
FDA explained that this is a legacy system that hasn’t been updated in years. For those who are not familiar with FDA’s history with information technology, in the early days, because the federal government didn’t have a professional core of IT specialists, scientists in different departments simply jerry-rigged their own databases. The scientists may have little to no formal training in database construction. As a consequence, they created these databases with different degrees of capability and foresight. Some of the systems work okay; some of them frankly do not. FDA has lots of these legacy systems lying around in areas where they haven’t chosen to allocate budget to improve the data management system.
Actually improving the CLIA database would not be very difficult. For starters, they could simply come up with different codes for concurrent and standalone that wouldn’t require any reprogramming of the database itself. FDA could also add the dates at which standalone requests were made in a new field that presumably wouldn’t be that difficult to program.
But taking the database as it exists today, FDA explained that in some cases, the CR with the earliest Effective Date in the CLIA Database with a specific Parent 510(k) number may in fact be a standalone CR and not a concurrent CR. The reason for this is that in some cases a new categorization is not needed for the concurrent CR, for example if the manufacturer/distributor name, trade name, analyte(s), and complexity have not changed from a previous categorization. Additionally, if a correction has occurred for a concurrent CR, the Effective Date in the CLIA Database will be the date the correction was completed and the applicant was notified, rather than the date of the original concurrent categorization decision.
That does mean that individual data points in the charts above are potentially not reliable. I have no way of gauging just how common these anomalies are. But this does appear to be the best available public data to analyze. Hopefully, FDA will at some point update this legacy system.
Interpretation
An immediately obvious implication of the first chart is that the folks in toxicology seem to take longer than everyone else. The data don’t tell us why that might be. Everywhere else, the averages seem to be in an acceptable range.
When we look at the raw data, however, we can see that while the averages appear to be acceptable, there are still potentially many FDA decisions that take longer than they should. Those outliers tend to be moderate complexity decisions, although there are also quite a few waived decisions as well. High complexity represent the fewest outliers. Remember that I pruned all of the extreme outliers over 300 days. So I have some greater confidence in these data in the under 300 day range being meaningful, despite the limitations described above.
Ironically in the COVID years of 2020 and 2021, there are a lot fewer outliers. I have no idea why that would be. It’s interesting that in 2021, the high complexity decisions appeared to take a bit longer than in prior years, but still frankly less time than the outliers from the prior years.
As always, I’m interested in what you glean from these visualizations.
In Full Disclosure
At the risk that my prior involvement might color my judgment, I thought I should disclose my previous activity in this area on behalf of a coalition of medical device manufacturers and diagnostic trade groups.
Initially, responsibility for regulating these products was divided among the FDA, CDC and CMS. In 1999, I drafted a petition seeking improvements to the process of making CLIA categorization decisions. At the urging of our coalition, the process for evaluating new technologies was shifted from the CDC to FDA on January 31, 2000. 64 Fed. Reg. 73561 (December 30, 1999). After that, I further worked with FDA to develop a more efficient process for making CLIA waiver determinations reflected in the application process, which as I already mentioned I excluded from this analysis.
I should also mention that in 1997, I worked with Congress in its passage of the FDA Modernization Act to clarify that an FDA determination that a product is simple enough to be used in the home means it’s also simple enough to be waived under CLIA.
* * * *
The Unpacking Averages® blog series digs into FDA’s data on the regulation of medical products, going deeper than the published averages. The opinions expressed in this publication are those of the author(s). Subscribe to this blog for email notifications.